







Insight
July 8, 2019
The Inconsistent Treatment of Biosimilars in Medicare and Medicaid
Executive Summary
- Biologics and biosimilars are some of the most expensive drugs due to high development costs and a limited pool of potential users.
- Biosimilars are often thought of as generic versions of biologic drugs, but federal policy is not consistent in its treatment of biosimilars.
- In its inconsistent treatment of biosimilars, federal policy appears to seek the greatest protection and choice for Medicare beneficiaries as well as the greatest cost reductions for the government and beneficiaries, though, sometimes that’s not always clear.
Introduction
References to biosimilars are quickly becoming more common in discussions of drug pricing. With more, and more expensive, biologic products coming to market, patients and policymakers are eager for generic-like versions to become available and provide a less expensive alternative. But biosimilars, by definition, are not technically generics, and the law does not always treat them like generics in terms of how they are reimbursed and the regulations governing their coverage on a prescription drug formulary. This paper provides some insight into those inconsistencies.
Background
The market for biologics has certain constraints. Biologics are complex molecules made from living organisms, and as a result, are much more expensive to develop and produce than small-molecule drugs made from chemicals. Further, biologics often treat rare diseases and, when they do, few patients use them (though, there are exceptions to this, with a good example being Humira, which was originally approved as a mass-market drug to treat rheumatoid arthritis (RA) and became the world’s best-selling drug before it was given multiple orphan drug designations for treatment of rare diseases).[1] This combination of high expense and a small natural market means their price is typically very high: In October 2016, MedPAC reported that biologics accounted for less than 1 percent of prescriptions, but 28 percent of spending in Medicare Part D.[2] All of the top 10 Medicare Part B drugs in terms of total spending in 2017 were biologic products and accounted for 46 percent of Part B drug expenditures despite accounting for only 7.5 percent of all claims.[3] Between 2006 and 2015, the prices of Part B-covered biologics grew 38 percent, on average, compared with a decline in prices of 18 percent for nonbiologics.[4] At the same time, the prices of Part D-covered biologics grew 195 percent, while the prices of all Part D-covered drugs grew just 67 percent.[5]
Insulin in 2014 made up the bulk of Medicare Part D spending on biologics. (Insulin is in fact a biologic, even though it is technically not currently classified as such; it will be on March 23, 2020.) The next largest share of spending on biologics in Part D was on drugs to treat inflammatory diseases, followed by drugs for Multiple Sclerosis.[6] In Part B, the top 10 biologics are used to treat cancer, RA, Chron’s disease, psoriasis, ulcerative colitis, and macular degeneration.
As of June 17, 2019, the Food and Drug Administration (FDA) has approved 22 biosimilars, although only 10 have been marketed in the United States and only 7 are commercially available, while more than 40 have been approved for use in Europe.[7] The limited selection might indicate that more options would lower prices, but even if more biosimilars were on the market, it’s not clear that significant savings would develop: It costs between $100-$250 million to develop a biosimilar product, compared with $1-$4 million to develop a generic drug.[8] This difference is largely because of the complexity of the approval process and the difficulty in proving a drug’s similarity to the reference product.[9]
Coverage
Regarding Medicare Part D’s coverage requirements, biosimilars are treated in a way that provides beneficiaries the greatest choice and protection; or at least they will be once biosimilars enter the Part D market.
In the context of Part D’s requirement for insurers to cover at least two drugs per class, biosimilars are not considered distinct from their reference product. Thus, covering only a reference biologic and its biosimilar will not satisfy the “two drugs per class” rule.
When it comes to “transition fill coverage,” however, the drugs are considered distinct. Rules require plans to offer beneficiaries coverage of a one-time 30-day supply of a drug if the beneficiary is already taking the drug and is losing coverage of that drug either because they are switching plans or the insurer is changing its coverage in the next calendar year.[10] An insurer may stop covering a brand-name drug or may place coverage restrictions on the drug (such as step therapy or prior authorization) if a generic version becomes available in order to encourage patients to take the less expensive medicine. The transitional fill coverage requirement is intended to help smooth the transition for beneficiaries on an existing prescription regimen. This requirement will not be satisfied by covering a biosimilar product if the beneficiary is taking the reference product, since the two are considered distinct in this context.
Similar to transition fill coverage, biosimilars are considered distinct from the reference product in the context of mid-year formulary changes. Insurers are allowed to stop coverage of a brand-name drug, move a brand-name drug to a higher tier, or add coverage restrictions to a drug if a generic drug becomes newly available; these are considered maintenance changes and require approval from the Centers for Medicare and Medicaid Services (CMS), but they are typically granted.[11] Dropping a brand-name biologic in favor of a biosimilar, however, is considered a non-maintenance change, since they are considered distinct drugs in this context, and CMS is less likely to approve this change.
Reimbursement
Medicare Part B does not treat biosimilars like either generics or brand-name drugs. For small molecule drugs in Part B, if a generic drug exists, then the reimbursement for that drug is the same whether a patient takes the brand-name or generic version. The reimbursement amount is based on the volume-weighted average sales price (ASP) of both the brand-name and generic versions of the drug, which reduces the reimbursement rate (relative to what it would be if based solely on the price of the brand-name drug) and therefore encourages use of the cheaper generic version. For single-source drugs and biologics, the reimbursement rate is 106 percent of the ASP of the drug. For biosimilars, a blended reimbursement formula is used, though the precise formula was changed through regulation, effective January 1, 2018. Originally, all biosimilars of a given reference product were reimbursed equally, with the base payment amount based on the ASP of all the biosimilars, but the add-on payment equal to the add-on payment for the reference product, as explained here. This provided a reimbursement amount that was less than that provided for the innovator drug (saving money for Medicare) while not disincentivizing providers from using the lower-cost drug (because the add-on payment was the same as that for the higher-priced drug). Critics complained, however, that biosimilars should not be reimbursed this way since one biosimilar may treat different or fewer indications than another biosimilar of the same product. In response to this criticism, CMS issued a new regulation in November 2017 that would provide a separate billing code and reimbursement amount for each biosimilar using each individual drug’s own ASP for the base payment amount and maintaining the use of the reference biologic’s ASP for the add-on payment.[12] This new reimbursement policy may lead to higher spending, though—at least initially, as the change reduces price competition among similar products. It is hoped, however, that the more favorable reimbursement will encourage more biosimilar market entrants, ultimately saving money by shifting market share away from the more expensive innovator products.
In Part D, until the Balanced Budget Act (BBA) of 2018, biosimilars were treated as generics in the coverage gap discount program (CGDP), meaning they were not required to pay the mandatory rebate that brand-name manufacturers were required to pay. With brand name drugs, the manufacturer’s discount is applied toward the beneficiary’s calculated out-of-pocket cost (known as true out-of-pocket (TrOOP) cost), as though the beneficiary paid the amount covered by the discount, accelerating the beneficiary through the coverage gap. For generics, in contrast, beneficiaries faced higher coinsurance rates for such drugs and also did not benefit from accelerated TrOOP calculations. The BBA changed this policy, and now biosimilars must pay coverage gap rebates. This change may not hurt biosimilars however, since the previous rules made biosimilars less favorable to insurers from an insurer liability perspective: During the gradual transition to close the coverage gap, insurers were required to cover a greater share of the cost of generics and biosimilars than they were for brand-name drugs, as shown here.
Despite the BBA’s changes in February 2018 regarding the coverage gap discount, in April 2018 CMS finalized a rule to treat biosimilars as generics when it comes to determining the cost-sharing amount for beneficiaries eligible for the low-income subsidy (LIS).[13] LIS beneficiaries are only required to pay between $1.25 and $3.40 for generic and biosimilar drugs and $3.80 and $8.50 for brand-name drugs.[14]
In Medicaid, biosimilars are treated as single-source brand-name drugs for purposes of determining their minimum required rebate amount under the Medicaid Drug Rebate Program.
The following chart summarizes how biosimilars are characterized for purposes of reimbursement under the various federal health insurance programs:
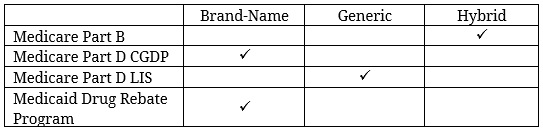
The differences in reimbursement policy in Medicare Parts B and D and Medicaid all seem aimed at reducing costs for patients and the government—whether by requiring the biosimilar manufacturers to pay larger rebates or providing patients and providers financial incentives to use biosimilars relative to brand-name products.
Conclusion
While biosimilars are sometimes treated like single-source, brand-name drugs and sometimes treated like generics, the one consistency seems to be that each decision is designed to increase choice and reduce costs. In terms of coverage, policies are aimed at giving patients the greatest amount of choice and protecting their access to medications. In terms of reimbursement, policies aim to reduce the government’s and patients’ costs, either directly through greater rebate requirements or indirectly by encouraging development and utilization of biosimilars over the brand-name reference product. Yet just as the biosimilar market is evolving, these policies could as well, and they are worth watching.
[1] https://khn.org/news/drugmakers-manipulate-orphan-drug-rules-to-create-prized-monopolies/?rel=0
[2] http://www.medpac.gov/docs/default-source/default-document-library/biosimilarsinmedicarepartd_oct16_pres_sec.pdf?sfvrsn=0
[3] https://www.cms.gov/Research-Statistics-Data-and-Systems/Statistics-Trends-and-Reports/Information-on-Prescription-Drugs/MedicarePartB.html
[4] http://www.medpac.gov/docs/default-source/data-book/jun18_databooksec10_sec.pdf?sfvrsn=0 (pg. 153, 182)
[5] http://www.medpac.gov/docs/default-source/data-book/jun18_databooksec10_sec.pdf?sfvrsn=0 (pg 181, 182)
[6] http://www.medpac.gov/docs/default-source/default-document-library/biosimilarsinmedicarepartd_oct16_pres_sec.pdf?sfvrsn=0
[7] https://biosimilarsrr.com/us-biosimilar-filings/, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6355057/, https://www.biosimilarscouncil.org/wp-content/uploads/2019/06/Biosimilars-Council-White-Paper-Failure-to-Launch-June-2019.pdf
[8] https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4031732/
[9] https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6355057/
[10] https://www.medicareinteractive.org/get-answers/medicare-prescription-drug-coverage-part-d/medicare-part-d-coverage/transition-drug-refills
[11] https://www.cms.gov/Medicare/Prescription-Drug-Coverage/PrescriptionDrugCovContra/Downloads/Part-D-Benefits-Manual-Chapter-6.pdf
[12] https://www.federalregister.gov/documents/2017/11/15/2017-23953/medicare-program-revisions-to-payment-policies-under-the-physician-fee-schedule-and-other-revisions
[13] https://www.centerforbiosimilars.com/contributor/brian-lehman/2018/04/cms-finalizes-policy-to-lower-the-cost-of-biosimilars
[14] https://www.ncoa.org/wp-content/uploads/part-d-lis-eligibility-and-benefits-chart.pdf
