







Insight
November 17, 2025
Primer: Prescription Drug User Fee Agreement
Executive Summary
- The Food and Drug Administration’s (FDA) user fee programs – including the Prescription Drug User Fee Agreement (PDUFA) – are five-year, negotiated agreements that pair industry fees with performance goals and public reporting, and collectively form the backbone of modern review capacity.
- Across successive program cycles, the user fee agreements have shortened and stabilized review timelines, expanded sponsor–FDA engagement, and funded critical capacity to support earlier approval and access to many innovations while strengthening postmarket signal detection and labeling updates; the reauthorization currently being negotiated may set clearer frameworks for artificial intelligence/machine learning and digital endpoints, stronger tools for initiating and enforcing confirmatory studies, continued workforce and IT modernization, smarter workload and fee calibrations, and greater transparency and global harmonization.
- This primer explains the history of PDUFA, what the latest agreement funds and expects, where the benefits are most tangible, and which policy choices will shape the next reauthorization.
Introduction
The Prescription Drug User Fee Act (PDUFA) is the backbone of the modern U.S. drug review system. Enacted in 1992 amid long backlogs and unpredictable timelines for drug approvals, PDUFA authorized the Food and Drug Administration (FDA) to collect fees from sponsors in exchange for meeting transparent performance goals. Each cycle, the FDA prepares a “commitment letter,” in which it specifies for sponsors review timelines (and the familiar “PDUFA date”), meeting frameworks that promote earlier scientific engagement, plans to recruit and train specialized reviewers, and upgrades to data standards, analytics, and IT. A statutory “trigger” guards the principle that user fees supplement, rather than replace, congressional appropriations.
This structure has delivered speed with predictability. Median first-cycle decisions (when new drug applications (NDAs) are approved on the first submission) are faster and more consistent; sponsors and clinicians can plan goal dates; and postmarket work – signal detection, labeling updates, risk management, etc. – has expanded alongside review capacity. Yet PDUFA’s prominence also fuels enduring debates: optics of industry funding, the risk that deadline pressure could erode late-cycle deliberation, and the need for timely confirmatory evidence when approvals rely on surrogate endpoints (a predictive, indirect measure, like a lab test or a physical sign) rather that clinical endpoints (a direct measure of how a patient feels, functions, or survives).
Recent cycles have emphasized evidence modernization – model-informed drug development, complex trial designs, and responsible use of real-world evidence – and chemistry, manufacturing, and controls (CMC) readiness to reduce late-cycle surprises. The current agreement also begins to clarify how digital health tools and artificial intelligence (AI)/ machine learning (ML) methods can be incorporated, with attention to transparency, validation, and lifecycle management. Looking ahead, priorities are converging around making innovation evaluation both faster and sturdier. Expect clearer expectations for AI/ML documentation, validation, and lifecycle change control; digital endpoints and decentralized trial operations moving from guidance to routine practice; earlier and deeper engagement on manufacturing readiness and quality maturity to avoid CMC-driven delays.
This primer explains the history of PDUFA, what the latest agreement funds and expects, where the benefits are most tangible, and which policy choices will shape the next reauthorization.
Why PDUFA Was Created
PDUFA emerged to solve a structural mismatch rather than a single policy failure. By the late 1980s, FDA’s review workload had outgrown its base of congressional appropriations. Application volume and scientific complexity were rising (oncology, antivirals, biologics), yet reviewer hiring, training, and IT were constrained by annual budget cycles. The result wasn’t just “slow reviews,” it was unpredictability: uneven staffing across disciplines, late-cycle information requests, and timelines that varied widely by division and product type. Sponsors struggled to plan development and launch, and patients and clinicians couldn’t reliably anticipate when important therapies would become available.
Predictable, multi-year resources could provide a source of stability, enabling the agency to underwrite hiring and training, modernize systems, and stabilize review teams. In return, FDA would commit to measurable performance goals, clearer meeting frameworks, and public reporting – creating accountability without compromising scientific standards. The five-year cycle was deliberate: long enough to plan hiring and IT roadmaps, short enough for Congress and stakeholders to recalibrate goals as science and workloads evolved. In short, PDUFA’s design paired durable capacity with enforceable predictability, converting an episodic, backlog-prone process into a managed system with known timelines, defined touchpoints, and incentives for higher-quality submissions.
PDUFA Fees
PDUFA fee collections flow through several pillars designed to align revenue with workload. First are application fees, paid when a sponsor submits a new drug application or biologics license application (BLA) requiring clinical data. These fees support the surge of intensive review work – disciplines such as clinical, biostatistics, clinical pharmacology, CMC, and labeling. Second are annual program fees, assessed on marketed prescription drug and biologic products. These sustain ongoing review capacity – postmarket safety signal evaluation, labeling updates, manufacturing changes, and supplements. Each reauthorization refines how fees are set and adjusted. Indexing mechanisms account for inflation, shifts in workload (e.g., more NDAs, more BLAs, more supplements), and strategic investments (e.g., IT modernization). A key guardrail is the appropriations “trigger:” user fees can only be collected if Congress maintains a specified baseline of budget authority. This prevents fees from supplanting taxpayer funding and preserves the principle that core public-health functions remain a governmental responsibility. FDA also manages carryover balances so hiring surges, IT upgrades, and multi-year initiatives can be planned without cliff effects.

Goals and Governance
PDUFA operates on a five-year cycle. Before each reauthorization, FDA holds public meetings to identify needs and priorities, negotiates with the regulated industry on a draft commitment letter, and invites public comment before submitting the final package to Congress. The commitment letter becomes the operational blueprint: performance goals for review timelines (standard versus priority), clock-start definitions, and goal-date reporting; meeting frameworks (e.g., Type A/B/C/D) with scheduling and response-time targets; and program enhancements such as reviewer hiring plans, training, data standards, and IT deliverables. Transparency is built in: FDA publishes performance reports tracking on-time actions, meeting timeliness, and staffing progress. The structure incentivizes early, frequent, and focused sponsor-FDA engagement, which tends to yield higher-quality submissions and fewer late-cycle surprises. The table below shows the evolution of PDUFAs over the years.
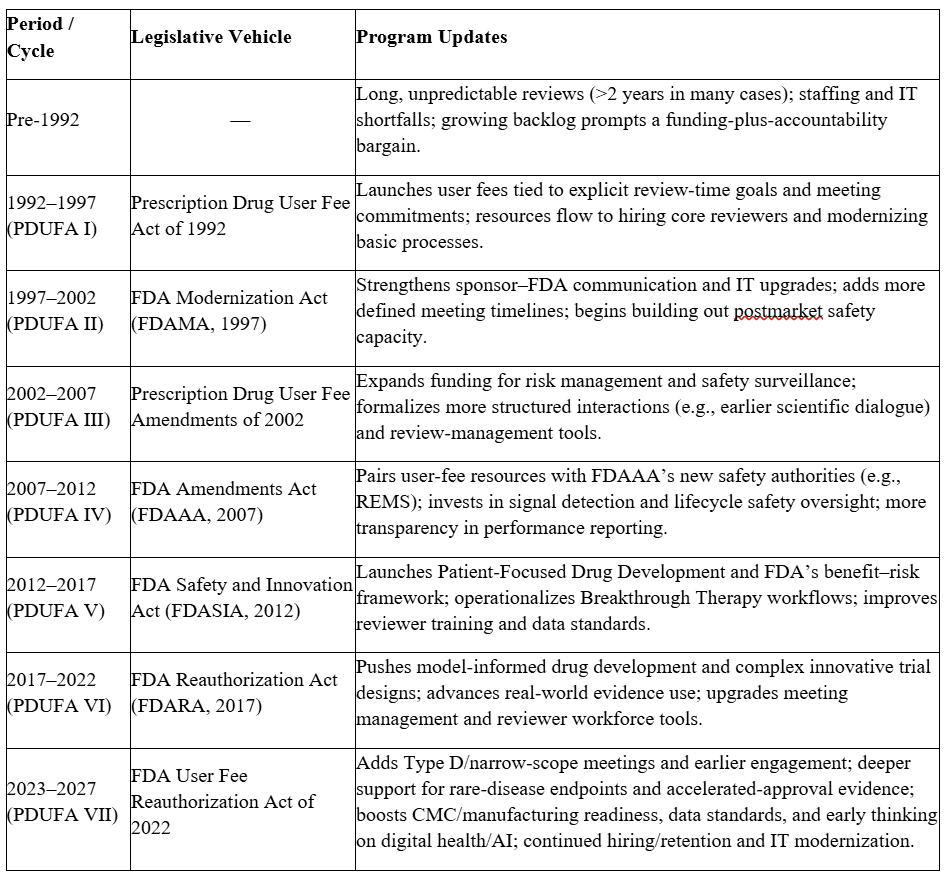
The Current Cycle: PDUFA VII
The current PDUFA cycle has emphasized three broad themes. First, the FDA and the industry agreed on earlier and better engagement, including expanded meeting options and clearer expectations for milestone interactions aim to resolve scientific issues upstream – whether around trial endpoints, exposure–response analyses, or CMC readiness – so that the final submission is stronger. Second, the FDA has emphasized evidence modernization, such as initiatives to support innovative trial designs, model-informed drug development, and thoughtful use of real-world evidence, especially in rare diseases where traditional trials can be infeasible. Finally, PDUFA ensured platform and workforce capacity through investments that target reviewer recruitment and retention, postmarket safety tools, and IT upgrades for submission intake, data standards, and analytics. Cross-cutting attention to manufacturing quality and supply resilience aims to surface CMC risks early and reduce last-minute approval delays tied to facilities or manufacturing capacity. FDA has also begun clarifying how digital health tools and AI/ML-enabled methods can be incorporated into submissions and assessments, with an emphasis on transparency, validation, and lifecycle management. The unifying theme is predictability: clear lane markers for sponsors and better-resourced review teams at FDA.
PDUFA Impacts
Across cycles, PDUFA has delivered faster and more predictable first-cycle decisions for many novel drugs and biologics. Goal dates – commonly called “PDUFA dates” – anchor planning for FDA, sponsors, clinicians, and patients, while adherence reporting creates accountability. Increased resources gathered from the authorized user fees have allowed FDA to stand up specialized review functions (e.g., pharmacometrics, safety analytics, informatics) that improve scientific rigor and consistency. Communication frameworks and meeting commitments encourage iterative dialogue; sponsors can align endpoints, trial populations, and CMC strategies before submitting a drug for review, which reduces deficiencies and rework. The United States has often been among the first markets to make innovative therapies available, particularly in oncology, rare diseases, and specialty areas – reflecting both scientific leadership and a well-resourced regulatory process. In the postmarket environment, PDUFA resources support signal detection, labeling updates, risk-management plans, and ongoing evaluation of benefit–risk as real-world use expands. Just as important, the program has normalized continuous improvement inside the agency – each cycle funds not only reviewer headcount, but also training, tools, and data standards that compound efficiency over time.
Themes in the Next Reauthorization: PDUFA VIII
Looking ahead to the next reauthorization, one should expect the integration of the priorities that the FDA commissioner and the larger Department of Health and Human Services have expressed. This could include two cross-cutting themes: using targeted pilot programs to trial policy before cementing it and confronting persistent workforce capacity and retention issues that affect review predictability.
Recent remarks from FDA leaders suggests the agency may lean more on pilots and flexible industry communications rather than immediate, formal guidance to explore emerging methods (e.g., complex innovative designs, model-informed development, digital endpoints) and to de-risk process changes before scaling them across divisions. It also remains to be seen whether the reauthorization negotiations will address policies related to drug costs – historically a “third rail” for FDA’s user-fee talks – while noting that the more realistic near-term priorities are pilot design, scope, and uptake.
FDA’s commitments on staffing, another large component of the PDUFA process, underscore major issues: The agency convened a public Hiring and Retention Assessment meeting in September 2025 (a PDUFA VII requirement), signaling that recruitment, pay flexibilities, training, and retention remain central to on-time reviews. Expect PDUFA VIII to carry forward measurable hiring and retention deliverables and transparent reporting, because reviewers – not just standardized timelines – are the binding constraint in several high-complexity areas.
PDUFA’s tradition of pilots offers a ready playbook. Prior cycles stood up programs such as Model-Informed Drug Development paired meetings, the Complex Innovative Designs pilot, and streamlined-review experiments – all mechanisms that test feasibility, evidence thresholds, and operations while giving sponsors structured agency feedback. Expect similar constructs in PDUFA VIII, potentially including rare-disease endpoint pilots that create earlier touchpoints on clinically meaningful outcomes. The comment docket already features proposals along these lines, reinforcing that pilots may be the practical vehicle for rare-disease evidence development in the next cycle.
Substantively, anticipate some continuity with the current cycle on AI/ML credibility and change control, digital endpoints and decentralized data capture, earlier CMC and manufacturing readiness to reduce late-cycle plant or quality surprises, and strengthened postmarket/accelerated-approval follow-through (clearer expectations for when confirmatory trials must be underway and how milestones are enforced). For example, expect a tighter, more explicit framework around AI and ML in both development and review. FDA has been laying the groundwork with credibility and validation concepts; the next cycle could fully integrate documentation standards, change-control expectations when models evolve, and reviewer-facing processes to assess algorithmic tools used in clinical, statistical, and CMC work. In parallel, digital health technologies may move from guidance into routine practice. That means clearer guardrails for digital endpoints, remote data capture, and decentralized or hybrid trial operations, particularly around endpoint validation, data integrity, and operational feasibility in real-world settings. Those priorities have been steadily elevated in FDA’s kickoff materials and guidance dockets and are well-suited to pilot-first approaches that generate shared learning before broad implementation.
Building on recent policy signals, expect firmer expectations for when confirmatory trials must be underway, more precise milestones for completing them, and stronger levers to ensure timely evidence submissions after accelerated approvals. Earlier facility engagement, quality-maturity expectations, and governance for model-enabled manufacturing and analytics will aim to reduce late-cycle surprises and strengthen supply resilience. Expect some focus on global alignment and supply resilience, which may include greater harmonization of data standards and inspection practices with peer regulators, and earlier engagement on manufacturing readiness to reduce approval-stage quality bottlenecks.
Underpinning all of this, the program will likely extend FDA’s data/IT modernization while continuing to invest in reviewer recruitment, retention, and training. Fee mechanics – such as how workload adjusters and carryover targets are calibrated – may also be fine-tuned to smooth hiring cycles and keep pace with scientific complexity.
Next Steps
By law, reauthorization follows a set sequence. Before negotiations with industry begin, FDA must: publish a Federal Register (FR) notice, hold a public meeting, keep a 30-day comment window, and post comments publicly. PDUFA VIII formally opened with FDA’s Federal Register notice on May 19, 2025, announcing a hybrid kickoff public meeting for July 14, 2025, and a 30-day post-meeting comment window through August 14, 2025.
During negotiations, FDA must consult with patient and consumer stakeholders at least monthly and post minutes of FDA-industry negotiation meetings within 30 days. After negotiations, FDA publishes draft recommendations (“commitment letter”) in the FR, takes another 30 days of comment, and holds a second public meeting before finalizing.
Through 2026, there will be ongoing negotiations and recurring stakeholder-consultation sessions; these signal emerging priorities, such as meeting frameworks, CMC and manufacturing readiness, digital/AI evidence, postmarket commitments. Two hard dates anchor the process. First, FDA must transmit final revised recommendations to Congress by January 15, 2027 – including a summary of comments and any changes. Second, PDUFA VII’s fee-collection authority sunsets on October 1, 2027; Congress must enact reauthorization legislation by then to avoid a lapse.
