







Weekly Checkup
October 31, 2025
Finally, a Drug Policy Focused on Competition
Welcome to the party, HHS.
On Wednesday, the Department of Health and Human Services (HHS) and the Food and Drug Administration (FDA) released draft guidance that squarely leans into competition by lowering unnecessary regulatory friction for biosimilars. The document, “Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Updated Recommendations for Assessing the Need for Comparative Efficacy Studies,” attempts to promote the development (and then adoption) of biosimilars for patients. This is a welcome step in the right direction to lower the cost of health care by promoting competition rather than implementing price controls.
At its most basic level, the draft guidance reframes the criteria for head-to-head clinical efficacy trials. FDA proposes that if a product’s lab-based comparative analytical assessment shows it is highly similar to the reference biologic – and developers provide an appropriate human pharmacokinetic study and an immunogenicity assessment – those costly comparative efficacy studies may be unnecessary. Instead, the FDA will judge applications on the “totality of evidence” with the most sensitive tests up front. The guidance also flags exceptions (e.g., some locally acting products) where comparative clinical work may still be useful. The intent? Reduce time and expense of biosimilar development without lowering regulatory standards, thereby speeding competition against high-spend biologics and helping payers and patients see savings sooner. FDA paired the guidance with messaging that this policy shift targets “unnecessary, resource-intensive” requirements while keeping safety and efficacy expectations intact.
As seen in the table below, there has not been a consistent trend in the approval of biosimilar products. While there are myriad reasons for this, the FDA is attempting to create some momentum.
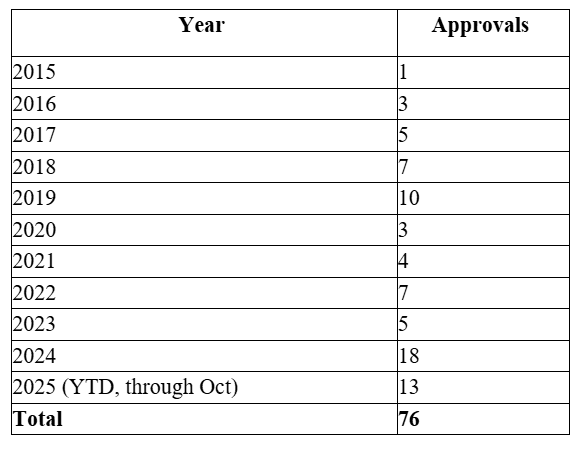
Source: Food and Drug Administration
This is welcome news for those advocating unleashing innovation and competition as key forces to bring down health care prices in the United States. The American Action Forum has long promoted competition as the cleanest, most durable way to lower drug prices. The strongest proof of this comes from what happens when generics and biosimilars are introduced. FDA’s own economic work shows a simple relationship: As soon as even a few generic competitors enter the market, prices fall, and further entrants drive them lower still. In 2022 alone, the cohort of newly approved generics generated an estimated $18.9 billion in savings within 12 months.
Biosimilars bring the same logic to biologics, where single-source spending is concentrated. The Biologics Price Competition and Innovation Act of 2009 (BPCIA) – enacted as part of the Affordable Care Act – created the statutory pathway for U.S. biosimilars. It amended the Public Health Service Act to add section 351(k), an abbreviated licensure route for products shown to be “biosimilar to” or “interchangeable with” an FDA-licensed reference product and set data/market-exclusivity periods and a patent-information exchange process.
HHS estimates that biosimilar competition lowered Medicare Part B spending on affected drugs by about 62 percent in 2023 versus a no-competition projection, with patients who used these products saving nearly $2,000 on average out-of-pocket that year. Independent oversight has reached similar conclusions. HHS’s Office of the Inspector General finds biosimilars have already reduced Part B costs for the program and beneficiaries while also highlighting headroom for still greater savings as uptake grows. A recent IQVIA report notes that biosimilars have saved the U.S. health care system $36 billion, and over the next decade, 118 biologics are expected to lose patent protection. This presents an approximately $234 billion market opportunity for biosimilars.
Full policy implementation is always more complicated than the announcement of any guidance, however, and this is doubly true here. First, the BPCIA sets out robust criteria for biosimilar approval and glossed over in the announcement was how FDA planned to align with the interchangeability provision in the BPCIA. While the FDA has some authority to determine interchangeability, the agency has long advocated congressional action; it appears to be a dramatic shift to simply decide it doesn’t need new authority now. Second, states will need to update pharmacy regulations to solve some interchangeability issues, given that state boards of pharmacy are chiefly responsible for prescription authority. Third, companies will still have to invest in the development and manufacturing of biosimilar products, and tough economic conditions brought on by market instability (including tariffs and trade relations) may lessen the feasibility of bringing biosimilars to market. Finally, as with most pharmaceutical policy discussions, the issue of intellectual property regulations will certainly play a role.
Still, the health care industry should be excited about the FDA’s instinct to promote competition rather than impose stringent, market-controlling regulations. If coupled with a coherent payment policy and continued attention to market frictions, the shift in regulatory stance could deliver broader, faster biosimilar entry – and with it, the kind of durable savings and access gains that price-setting can’t produce. That’s a welcome pivot toward market-based policy at a moment when patients need every dollar to count.
