







Research
August 22, 2017
Primer: FDA User Fees
Executive Summary
The current Food and Drug Administration (FDA) User Fee Acts for medical products were set to expire on September 30, 2017, but new agreements were signed into law on August 18, 2017, extending the programs’ authorization for another five years. The law authorizes four user fee acts to help fund the FDA: the Prescription Drug User Fee Act (PDUFA); the Medical Device User Fee Act (MDUFA); the Generic Drug User Fee Act (GDUFA); and the Biosimilar User Fee Act (BsUFA). The FDA collects user fees from drug, device,biologic companies and uses them to fund its technology and workforce to speed review of industry product applications, and to more quickly bring new products to market. While these user fees are now critical to the FDA’s operation, they did not always enjoy the widespread support they have today.
Overview
The government has a long history of trying to implement user fees to increase available funds for public organizations. However, this had always been unpopular in the drug industry and as of 1983, neither the FDA nor the pharmaceutical industry supported FDA user fees.[1] The FDA opposed the imposition of user fees due to concerns about the effect such fees would have on industry innovation, consumer costs, and the integrity of the FDA. It argued that user fees would discourage innovation from small companies with less revenue, and that larger pharmaceutical companies would simply pass the cost of user fees along to the consumer in the form of increased drug prices. An additional concern was that industry funding of a public organization would place the FDA in the back-pocket of pharmaceutical companies and would make it difficult for the FDA to remain autonomous. Industry mainly opposed user fees because it did not want to pay additional costs for a mandatory service.[2]
Despite initial industry opposition, long review processes impede prescription drug manufacturers’ ability to recoup their research and development costs. Innovator prescription drugs receive a 20-year patent period and 12 years of market exclusivity during which the companies can recoup investments before competition comes on the market. However, the long FDA review process consumes valuable years of the drug’s patent, pressuring industry to move its drugs to market as quickly as possible. Eventually, the backlog of drug applications at the FDA became large enough that the FDA and the industry agreed that user fees would benefit them both. In 1992, Congress authorized PDUFA, the first user fee agreement.[3]
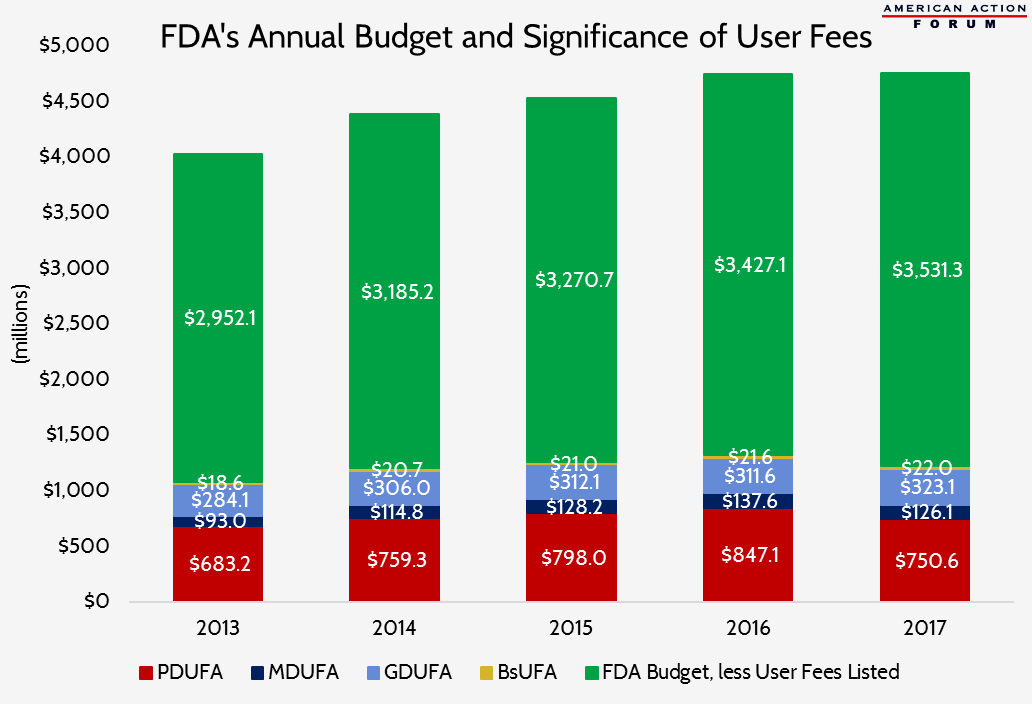
Over time, user fees have played an increasingly significant role in FDA’s budget. For the past several years, user fees have accounted for more than 40 percent of FDA’s overall budget, growing at a much faster pace than appropriations and becoming increasingly vital to the FDA’s ability to accomplish its mission.[4] From FY2012 to FY2016, appropriated funds increased by 9 percent while user-fee revenue increased more than 50 percent.[5] User fees for prescription drugs and medical devices have increased substantially since the first PDUFA agreement was signed twenty-five years ago.
Total User Fees Collected During Each Period ($ in millions)
| 1993-97 | 1998-2002 | 2003-07 | 2008-12 | 2013-17 | |
| PDUFA | $328.8 | $680.2 | $1,435.9 | $2,848.5 | $4,009.8 |
| MDUFA | $143.3 | $315.4 | $631.1 | ||
| GDUFA | $1,555.2 | ||||
| BsUFA | $92.6 |
The following chart shows the significance the prescription drug and medical device user fees play in FDA’s annual budget. These four user fees alone have accounted for 27 percent of the FDA’s budget over the past 5 years.
PDUFA
Goals
PDUFA was authorized as a compromise between the FDA and the industry and had four tenets: fees were additive to—not in place of—FDA funding; funds were only used for review of drugs and biologics; fees were reasonable; and fees were paid with the assumption that Congress and the FDA would research and improve the drug-review process.[6] These principles ensured that the FDA would have sufficient funds to accomplish its main goals, which were to clear the backlog of new drug applications (NDAs) and biologics license applications (BLAs), improve the infrastructure of the review process, and decrease the time that prescription drugs spend in review.
Structure
PDUFA authorized an application fee, an establishment fee, and a product fee for prescription drug companies. The application fee is charged to pharmaceutical companies for every NDA or BLA they submit. The establishment fee is an annual charge to pharmaceutical companies for every facility that manufactures a prescription drug or biologic on the market, which must be regularly inspected by the FDA. Industry companies also pay a product fee for each prescription drug that is sold on the market. In exchange for the FDA receiving the user-fee funds, it is expected to meet the goals upon which the industry and FDA agree. The goals differ between standard review products and priority review products, with priority given to drugs that dramatically improve the current treatment for an illness. Additionally, PDUFA contains requirements that 1) Congress must appropriate funds to the FDA equal to or greater than appropriations in 1992 and 2) the FDA must spend an equivalent proportion of appropriations on NDAs and BLAs as they did in 1992.[7] These provisions ensure that the user fees are additive to the FDA’s funding instead of a replacement for the funding and that the FDA uses that money to improve the review process. After five years, PDUFA expires and must be reauthorized in order for the FDA to receive user-fee funds in subsequent years.[8] This “sunset clause” forces the FDA and industry to continuously discuss and reassess the terms of the agreement and incentivizes the FDA to uphold its end of the deal.
Financing
During PDUFA I, the FDA collected over $328 million in user fees, which is divided equally among the three fee categories. Funding generally increased yearly from 1992 to 1997 as the 1992 baseline was adjusted for inflation and fluctuations in government discretionary spending.[9] PDUFA funds were used to hire nearly 700 employees during the five-year period, providing the FDA with the workforce necessary to meet its performance goals.[10]
Reauthorization
PDUFA mainly accomplished its goals and had widespread support by the time it expired in 1997. From 1993 to 1997, the average approval time for an NDA decreased from 27 months to 14.8 months.[11]
Under the FDA Modernization Act (FDAMA) of 1997, PDUFA II was authorized for the next five years. It set new goals to decrease approval time of NDAs and BLAs and planned to reach a decision on 90 percent of applications within 10 months as opposed to the 12 months the FDA had under PDUFA I. Additionally, the FDA committed to decreasing the amount of time drug manufacturers had to wait to meet with the FDA regarding the manufacturing of their drugs.[12]
In 2002, PDUFA III was authorized, again with wide public support for the bill. PDUFA was credited with helping to get several lifesaving cancer drugs on the market quickly, in as short as four months. However, PDUFA III had to address new challenges as the FDA reviewed more innovative drugs and faced an increasing workload and demand for NDA and BLA approval. As a result of the increased volume of applications, application fees tripled while total prescription drug user fees doubled during the five-year period of PDUFA III.
Concerns about post-market drug safety caused PDUFA III to include measures to expand the FDA’s scope of activities for which user fees could be used. Several drugs were pulled from the market for safety reasons, raising concerns that PDUFA had increased drug approvals at the expense of drug safety. The number of drugs withdrawn from the market had not, in fact, significantly changed since PDUFA was enacted.[13] Nonetheless, the controversy prompted Congress to allow the FDA to use user-fee funds for post-market safety studies. It also led Congress to require more safety guidelines, such as post-market safety responsibilities, but failed to give additional funding, resulting in financial strain on the FDA, and additional hikes in user fees.[14]
PDUFA IV was renewed in 2007. During this time, the FDA struggled to meet the post-market safety regulations due to insufficient funding of post-market research. The FDA also had several high-profile drug safety issues which did not increase faith in the agency. Once again fees increased, totaling nearly $3 billion by the end of PDUFA IV’s five-year period, and the FDA was granted expanded use of those fees.
The Food and Drug Administration Safety and Innovation Act (FDASIA) of 2012 authorized PDUFA V for an additional five years. Under PDUFA V, the FDA aimed to incorporate patient input into the drug approval process and improve the infrastructure of the drug application process and drug safety monitoring.[15] The FDA also intended to update its technology and knowledge of biomarkers and pharmacogenomics so it would be better equipped to review innovative research.[16] Additionally, the FDA committed to modernizing its technology and processes for several programs, including safety-tracking technology, benefit-risk assessment programs, and NDA, BLA, and investigational new drug (IND) application processes.
MDUFA
Structure
MDUFA was first established as the Medical Device User Fee and Modernization Act (MDUFMA) in 2002 and applies to the approval process of medical devices. Like prescription drugs, medical devices must be approved by the FDA and manufacturers are similarly required to perform clinical trials and show a device’s safety and efficacy. Components of the approval process for medical devices include premarket approvals (PMAs), product development protocols (PDPs), BLAs, some supplements, and premarket notification 510(k)s. PMA is a rigorous testing process that the FDA requires for life-sustaining devices or devices that are high-risk and with which it is unfamiliar. Because medical device companies must run extensive clinical trials, PMAs are very expensive, costing on average $94 million.[17] A PDP is a type of PMA for devices that use technology with which the FDA is familiar and has been proven safe. Prior to developing the drug, the manufacturer and the FDA agree upon what procedure and testing results would be acceptable, so when the device is deemed “complete” it is essentially automatically approved. A 510(k) is a process in which the medical device company compares safety and efficacy of its device to another similar model that has already been FDA approved.[18] If the manufacturer can show that the device is similar in safety and efficacy to the already-approved version, then its device’s approval is fast tracked. This method, costing around $31 million[19], is also substantially less expensive than a PMA.
Goals
Similar to PDUFA, MDUFMA established performance goals and timelines in order to increase transparency of the FDA review process. MDUFMA made a significant improvement to the review process of medical devices by allowing third-party inspections of medical devices.[20] While the FDA still implemented strict guidelines for these companies, it allowed FDA approval of innovative devices that the agency does not necessarily have the resources or knowledge to adequately assess. MDUFMA also established a new premarket submission called the premarket report, which is a type of regulatory requirement for reprocessed, single-use devices that is supposed to improve safety through labeling of single-use devices.[21]
Financing
MDUFMA was scheduled to raise $35 million in fees during the 2002-2007 period.[22] The fees are adjusted for inflation, changes in the workload, and requirements for the Department of Health and Human Services to adequately fund retirement packages for FDA employees. Fees are also tailored differently for small businesses, which tend to face fewer regulations and charges in order to foster competition in the medical device market.
Reauthorization
In 2007, Congress authorized MDUFA II, which included stricter goals for the FDA and increased user fees from the industry. Over the five-year period, MDUFA II was expected to collect $287 million from device companies, up from $35 million during 2002-2007.[23] It also charged the FDA with returning 50 percent of expedited PMAs and supplemental applications within 180 days and 90 percent of expedited PMAs and supplemental applications within 280 days. Ninety percent of 510(k) applications are to be returned in 90 days and 98 percent of 510(k) applications are to be returned in 150 days. MDUFA II helped small device manufacturing companies by decreasing user fees for small businesses and eliminating user fees for a small business’s first PMA.[24] According to a report from the Congressional Research Service, user fees paid by small businesses generally increased after MDUFA II, indicating that MDUFA II’s assist to small businesses led to small companies submitting more devices for review.[25]
When MDUFA II expired in 2012, Congress reauthorized MDUFA III, making alterations to the legislation that increased user fees, expanded the FDA workforce, increased FDA performance goals, and streamlined the application process. MDUFA III authorized $595 million in user fees, which enabled the FDA to hire approximately 200 new employees. These resources helped the FDA meet its performance goals, decreasing review time and increasing the number of applications evaluated.[26] MDUFA III also reauthorized the third-party review provision of 510(k) submissions, simplified the preauthorization process of applications, and required the FDA to submit more frequent progress reports.[27]
BsUFA and GDUFA
BsUFA and GDUFA are the most recent user fee acts, authorized by Congress in 2012. Their purpose is to increase competition among biologics and pharmaceuticals by improving the review process of biosimilar products and generic drugs.
BsUFA and GDUFA were signed into law as a part of the Food and Drug Administration Safety and Innovation Act. BsUFA applies to products that are biosimilar versions of biological products, which include products like allergenic extracts, tissue transplants, and gene therapies.[28] To receive FDA approval, manufacturers submit a 351(k) BLA proving that the biological product is “highly similar” to the approved product and submit results of animal studies and clinical studies showing safety and efficacy of the product.[29] BsUFA user fees fund the review of marketing applications and allow the FDA to hire additional employees for the review of biosimilars. BsUFA is beneficial to consumers because it helps put biosimilar products on the market faster, which gives consumers alternatives to high-cost biologics.
GDUFA significantly altered the drug market because prior to GDUFA, the FDA only collected user fees for NDAs, not generic drug applications. GDUFA authorized user fees for all abbreviated new drug applications (ANDAs), prior approval supplements to ANDAs, drug master files, annual facility fees, and a one-time fee for pending ANDAs to help clear backlogged applications.[30] The FDA plans to collect $299 million annually from generic drug user fees. Additionally, GDUFA simplifies the hiring process at the FDA for any GDUFA-related positions.[31]
FDA Reauthorization Act of 2017
All four of these user fee acts were set to expire at the end of this fiscal year. During the bill mark-ups, both the House Committee on Energy and Commerce and Senate Committee on Health, Education, Labor and Pensions (HELP) generally refrained from supporting unnecessary or controversial amendments, such as drug importation legislation, because they could have potentially prevented quick passage, though provisions related to encouraging generic drug development and approval were supported on a bipartisan basis.
The FDA Reauthorization Act of 2017 was signed into law on August 18, 2017. The legislation reforms the PDUFA fee structure, replacing the equal division of fees, such that 20 percent of fees will come from application fees and 80 percent will come from program fees; supplemental application fees and facility fees are eliminated. These changes are intended to provide greater predictability as to the amount of revenue that will be collected under these agreements each year. A new user fee for “de novo” medical device classification requests will be created. A generic drug applicant program fee will be established, which will account for 35 percent of the revenue for generic drug user fees and will be based on the number of ANDAs the applicant has approved; the fee for prior approval supplements will be eliminated. For the first time, an independent fee structure for biosimilars will be implemented, including an Initial Biosimilar Development Fee which will be assessed the first year a manufacturer begins clinical trials; an Annual Biosimilar Development Fee for subsequent years of the development process; a Biosimilar Program Fee for approved biosimilars; and an Application Fee for new biosimilar applications. The legislation also reauthorizes several programs that are designed to simplify and expedite the regulatory process for the development of drugs and devices that aid patients with rare diseases.
[1] https://www.fda.gov/aboutfda/whatwedo/history/overviews/ucm305697.htm#P3
[2] Ibid.
[3] Ibid.
[4] https://www.hhs.gov/about/budget/fy2017/budget-in-brief/fda/index.html
[5] https://fas.org/sgp/crs/misc/R44576.pdf
[6] https://www.fda.gov/aboutfda/whatwedo/history
[7]https://www.everycrsreport.com/files/20021007_RL31453_07f3e8333d43cbe71bab457740cc80e78b3a7372.pdf
[8] https://www.congress.gov/bill/102nd-congress/house-bill/5952/text
[10]https://www.fda.gov/RegulatoryInformation/LawsEnforcedbyFDA/SignificantAmendmentstotheFDCAct/FDAMA/ucm089179.htm
[11] Ibid.
[12] https://www.fda.gov/ForIndustry/UserFees/PrescriptionDrugUserFee/ucm174507.html
[13]https://www.forbes.com/sites/theapothecary/2014/08/08/faster-fda-approvals-have-not-caused-more-drug-safety-problems/#542ee157cf78
[14] https://www.fda.gov/aboutfda/whatwedo/history/overviews/ucm305697.htm#P3
[15]https://www.fda.gov/downloads/ForIndustry/UserFees/PrescriptionDrugUserFee/UCM270412.pdf
[16] https://www.fda.gov/aboutfda/whatwedo/history/overviews/ucm305697.htm#P3
[17] https://www.meddeviceonline.com/doc/are-you-sure-you-know-the-best-regulatory-pathway-for-your-new-medical-device-0001
[18] http://www.pmipreclinical.com/glp-difference-pma-510k-paths/
[19]https://www.meddeviceonline.com/doc/are-you-sure-you-know-the-best-regulatory-pathway-for-your-new-medical-device-0001
[20] https://www.fda.gov/ForIndustry/UserFees/MedicalDeviceUserFee/ucm109105.htm#1
[21] https://www.fda.gov/ForIndustry/UserFees/MedicalDeviceUserFee/ucm109105.htm#1
[22] https://www.fda.gov/ForIndustry/UserFees/MedicalDeviceUserFee/ucm109105.htm#1
[23] https://www.fda.gov/ForIndustry/UserFees/MedicalDeviceUserFee/ucm109317.htm
[24] https://www.fda.gov/ForIndustry/UserFees/MedicalDeviceUserFee/ucm109319.htm
[25] https://fas.org/sgp/crs/misc/R44517.pdf, Appendix A.
[26]https://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/PerformanceReports/UCM537819.pdf
[27] https://www.fda.gov/RegulatoryInformation/LawsEnforcedbyFDA/SignificantAmendmentstotheFDCAct/FDASIA/ucm313695.htm
[28]https://www.fda.gov/RegulatoryInformation/LawsEnforcedbyFDA/SignificantAmendmentstotheFDCAct/FDASIA/ucm311121.htm
[29]https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/UCM526935.pdfa
[30]https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm316671.pdf
[31] https://www.fda.gov/downloads/forindustry/userfees/genericdruguserfees/ucm282505.pdf
