







Weekly Checkup
May 1, 2026
RAPID Coverage, But Questions Abound
The Food and Drug Administration (FDA) and the Centers for Medicare and Medicaid Services (CMS) announced the Regulatory Alignment for Predictable and Immediate Device (RAPID) coverage pathway, an attempt to coordinate approval and coverage processes so that Medicare beneficiaries can access certain breakthrough medical devices more quickly after FDA authorization. FDA evaluates whether a device may enter the market; CMS determines whether Medicare will cover it. When those processes operate on disconnected timelines, patients may wait months or years between regulatory authorization and meaningful access. RAPID is intended to narrow that gap. But while the RAPID pathway may be a solution to some challenges, implementation could lead to the inappropriate convergence of approval and coverage decisions, and innovation incentives may erode.
The most attractive part of RAPID is its attempt to make Medicare coverage more predictable. Designed for certain FDA-designated Class II and Class III Breakthrough Devices, its basic premise is sensible. Under the pathway, CMS would issue a proposed national coverage determination on the same day an eligible device receives FDA market authorization, triggering the statutorily required 30-day public comment period. CMS and FDA say this could allow national Medicare coverage and payment as soon as two months after market authorization, compared with roughly a year or more under the current pathway. For manufacturers, that predictability matters. For patients, especially those with serious conditions and limited alternatives, the coverage delay after FDA authorization can be more than an administrative inconvenience; it can be the difference between theoretical innovation and actual treatment.
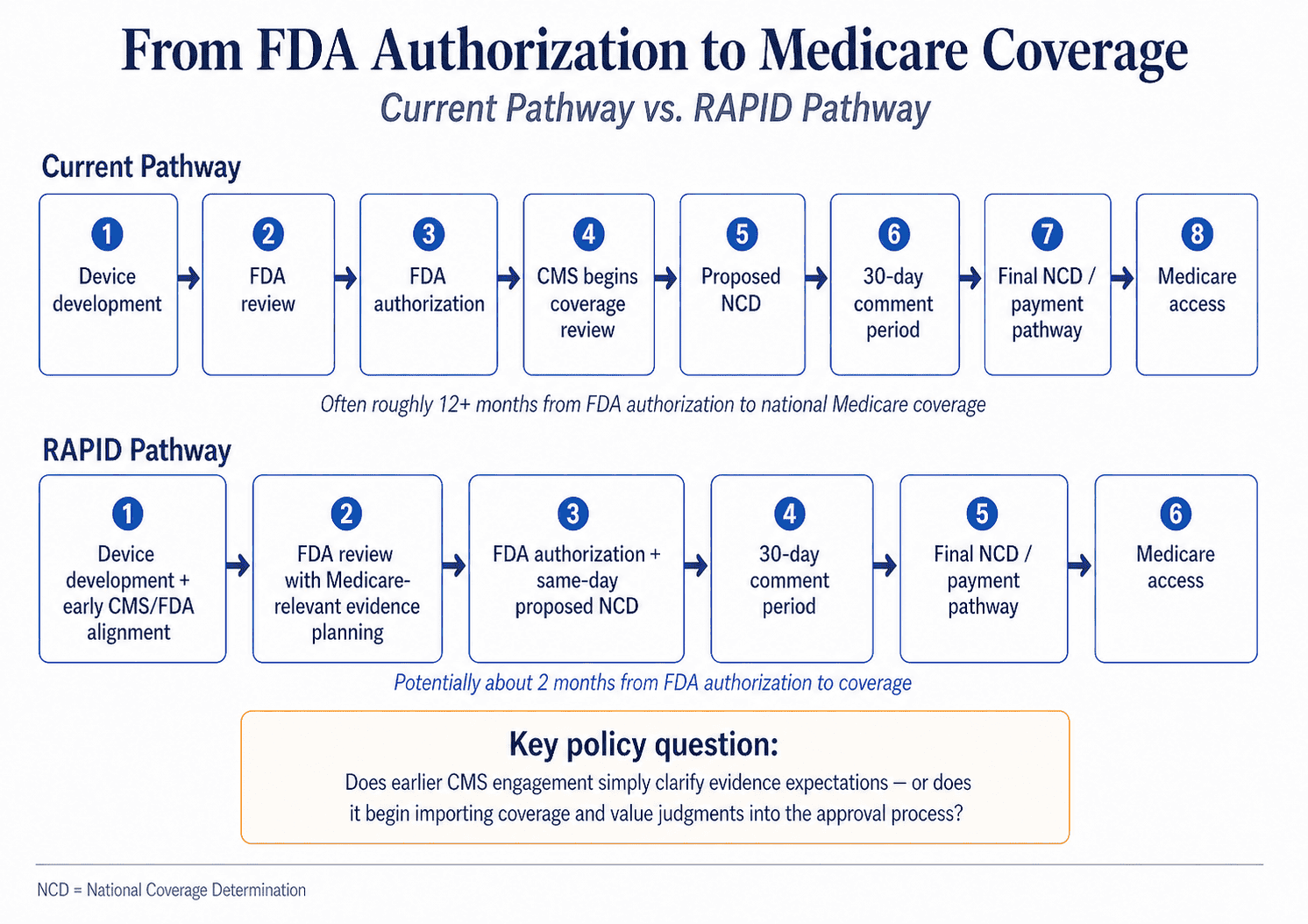
In that sense, RAPID represents a potentially positive example of government coordination. CMS and FDA describe the pathway as allowing the agencies to work with innovators earlier in the technology development lifecycle so that evidence generated for FDA review can also support Medicare coverage decisions. RAPID would link manufacturers to CMS experts early in development to understand what clinical outcomes are most relevant for Medicare beneficiaries. Eligible devices must also be the subject of an investigational device exemption study that enrolls Medicare beneficiaries and studies clinical health outcomes agreed upon by FDA and CMS. This may be a more coherent system than one in which innovators satisfy one agency only to discover, much later, that another agency has different expectations.
But the very feature that makes RAPID promising also makes it risky. The agency alignment may sound like mere coordination, but it also moves CMS closer to the front end of medical product development; this could change the traditional innovation incentives that currently guide the private market. Once CMS is involved in shaping the evidence package, it may become harder to preserve the distinction between regulatory approval and coverage judgment. FDA’s job is to determine whether a device is safe and effective under the applicable statutory standard. CMS’s job is to decide whether an item or service is reasonable and necessary for Medicare coverage, within a Medicare benefit category. Those are related questions, but they are not the same question.
CMS should not ignore evidence, but neither is Medicare required to cover every product merely because FDA has authorized it. Coverage decisions inevitably require judgments about patient population, clinical setting, real-world applicability, and whether an intervention improves outcomes for beneficiaries. The concern is that early CMS involvement could change private-sector incentives and gradually convert FDA-facing development into a quasi-health technology assessment (HTA) process, in which innovators are pushed to prove not only safety and effectiveness but also that a product delivers the right kind of “value” to federal health programs.
That would be a meaningful shift. The United States has historically resisted a centralized HTA model in which government uses comparative value judgments to determine whether technologies are worth covering. RAPID does not explicitly create such a system. Yet its design raises the question of whether coverage expectations will begin influencing product development before FDA has completed its independent review. If the evidence CMS wants is limited to Medicare-relevant clinical outcomes, that is appropriate. If it evolves toward judgments about whether the device is sufficiently cost-effective, budget-neutral, or valuable to the program, and deserves approval, the pathway could become a backdoor value assessment regime.
Fidelity to original program intent is particularly important because Breakthrough Devices are aimed at serious unmet needs. These are precisely the areas where evidence may be developing, patient populations may be small, and uncertainty may be unavoidable. The efficient coverage pathway that RAPID envisions should not become a way to delay or deny access that FDA wants to grant, but that CMS determines doesn’t reflect the appropriate value for coverage compared to the alternatives. Nor should breakthrough device innovators face a mercurial approval-and-coverage environment in which the evidentiary target shifts as different agencies – or their leaders – enter the process.
RAPID should therefore be judged by whether it simplifies review while preserving clear institutional boundaries. Earlier agency alignment can reduce duplication, clarify expectations, and shorten the access gap after FDA authorization. But CMS should not become a shadow regulator inside the FDA process, and FDA should not allow coverage considerations to reshape its scientific review. The RAPID pathway’s promise lies in making government work better together. Its danger lies in making the government work too much like a single gatekeeper for access.
